
本文是全系列中第6 / 7篇:生物信息学笔记
- 生物信息学笔记1
- 生物信息学笔记2:主机及环境配置
- 生物信息学笔记3:练手序列的准备
- 生物信息学笔记4:hisat2的下载安装及环境配置
- 生物信息学笔记5:用hisat2软件包建立基因组index
- 生物信息学笔记6:hisat2比对生成sam文件
- 生物信息学笔记7:对sam文件重新排序
在上一步,我们建立了index,可以在运行index命令的目录(我是在dnaseq\chrX_data\third\index,这个dnaseq目录是win10和linux的共享文件夹,关于该共享文件夹可能有些人可能还会有点弯路要走,我一开始是在linux下面找不到这个共享文件夹,这个后面如果有时间我会专门写一篇)下找到相应的文件如下图(里面包括了chrX.exon和chrX.ss这两个从gtf文件中提取的,以及剩下的就是index文件,有1-8,但是在比对时用chrX_tran代表):
然后,我们进入samples目录(就是我们下载了序列的目录),如下图:
注意了,上面图片示例都是在win10下截图的,只是为了更好理解,实际这一步还是在linux下操作比较方便,因为接下来要建立一个批处理文件samAA.sh是在linux下运行的。如下图:
建立方法,在linux,samples文件夹,输入命令:
touch samAA.sh
然后编辑该sh文件:
nano samAA.sh
在编辑其中输入命令:
for i in *1.fastq.gz;
do
i=${i%1.fastq.gz*};
nohup hisat2 -p 1 --dta -x /mnt/hgfs/dnaseq/chrX_data/third/index/chrX_tran -1 ${i}1.fastq.gz -2 ${i}2.fastq.gz -S /mnt/hgfs/dnaseq/chrX_data/third/sam-dta/${i}align.sam 2>/mnt/hgfs/dnaseq/chrX_data/third/sam-dta/${i}align.log &
done
保存退出后,在该文件夹下直接输入“sh samAA.sh”运行循环比对(多条序列比对)。
这个命令为什么这么写:
for语句第一行是要取出该目录下序列的文件名,就是1.fastq.gz前面的名字,比如“ERR188428_chrX_”,
do 开始循环,只要存在这样的i就继续执行,以其中循环一遍为例,比如让i=ERR188428_chrX_,
nohup开始在后台运行比对,比如把log保存在“/mnt/hgfs/dnaseq/chrX_data/third/sam-dta/ERR188428_chrX_align.log”里面,这样可以在文件里面查看是否成功,如果不成功问题提示如何。
hisat2 语句中“-p 1”是指单一进程运行比对,可以跟进电脑配置自行调整;
“--dta”是指输出的文件给stringtile继续使用;
“-S”注意这里是大写S,千万注意,这是指定位置输出比对好的sam文件,不要搞成小写s。
好了,比对结果如下图(看一下目录名,和命令行比较):
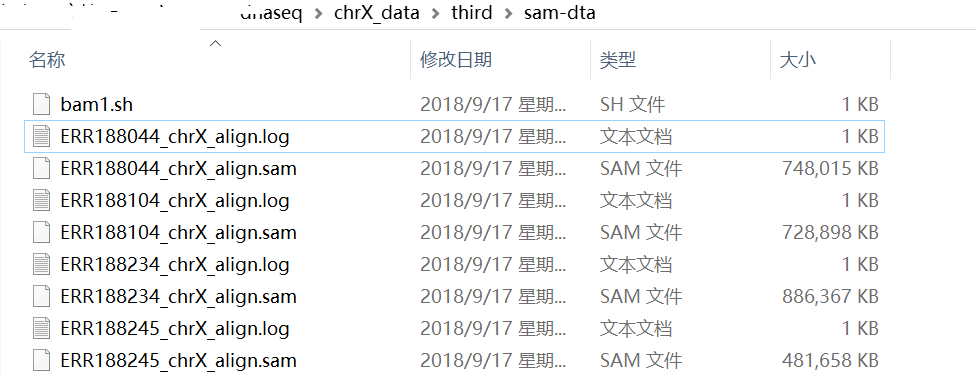
解释一下:命令行中有“/mnt/hgfs/”,后面才跟上dnaseq目录(这个是两个系统的共享目录),这个是因为linux下面的共享目录要挂载在/mnt/hgfs/下面。
因为比对时随机的,如果你用notepad++打开sam文件会发现里面的内容每次都是不一样的(但总大小一样,因为随机比对,顺序每次都不同),所以sam文件比对好以后还要进行排序,今天就到这里了。